Tensor decomposition methods in quantum chemistry
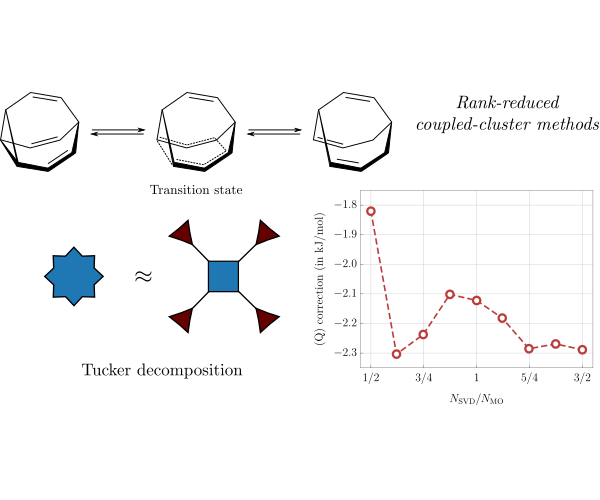 |
Tensors, understood as multi-dimensional arrays of numbers, are ubiquitous in quantum chemistry. For example, in coupled cluster theory
multi-dimensional tensors are used to represent the electronic wavefunction in terms of excited-state determinants. Storage and manipulation
of these tensors is largely responsible for unfavorable cost of the calculations. Our work focuses on applying tensor decomposition
techniques to the problematic tensors appearing in the electronic structure theory. This enables to extend the range of applicability
of the familiar quantum-chemical methods to much larger systems without significant impact on the accuracy of the results. The advantage
of the tensor decomposition methods is their universal character, as well as strict error control. As a result, they can be applied to
a variety of problems without additional assumptions such as spatial locality. We are currently working on extending this formalism to, e.g.
determination of molecular excited states and time-dependent phenomena.
Representative publications: M. Lesiuk, J. Chem. Theory Comput. 16, 453-467 (2020)M. Lesiuk, J. Chem. Theory Comput. 17, 7632-7647 (2021) M. Lesiuk, J. Chem. Theory Comput. 18, 6537-6556 (2022) |
|---|
Accurate ab initio calculations for small systems
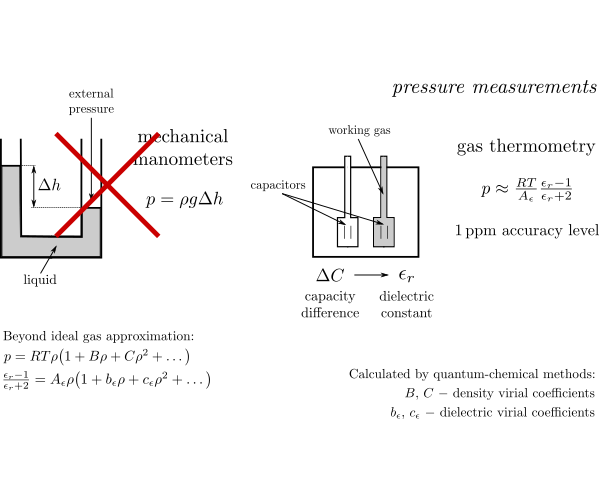 |
Accurate first-principles calculations for small atomic and molecular systems are a constant challenge for theoretical and computational
chemists. To match the accuracy standards of modern experimental techniques, new electronic structure methods have to be developed.
Our work in this area in largely inspired by recent advances in metrology, in particular by the development and refinement of experimental techniques
such as dielectric constant gas thermometry or refrative index gas thermometry. These methods establish a new standard for measuring fundamental
quantities such as pressure, and will replace older standards based on mechanical devices. However, gas thermometry experiments require a
significant theoretical input. In particular, to take into account deviations from the ideal gas model, the so-called pressure and dielectric
virial coefficients are needed. Our work concentrates on developing new theoretical methods to compute this quantities and on applying them
to systems relevant for the experimentalists.
Representative publications: J. Lang, M. Przybytek, M. Lesiuk, and B. Jeziorski, J. Chem. Phys. 158, 114303 (2023)M. Lesiuk and B. Jeziorski, Phys. Rev. A 107, 042805 (2023) P. Czachorowski, M. Przybytek, M. Lesiuk, M. Puchalski, and B. Jeziorski, Phys. Rev. A 102, 042810 (2020) |
|---|
New organic materials for light-emitting diodes
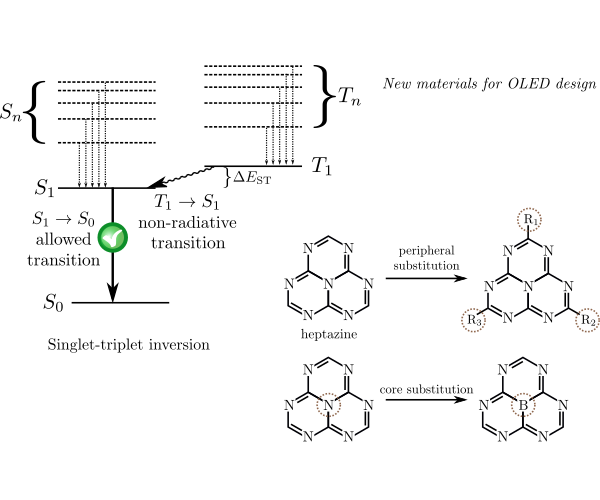 |
Organic light-emitting diode (OLED) is a device which emits light in response to an electric current and where electroluminescent layer comprises an organic compound responsible for the emission. The applications of OLEDs are widespread and nowadays they are found in numerous pieces of modern technology, such as mobile phones and digital cameras, among others. Therefore, the design of new electroluminescent materials for OLEDs is an endevour of great academic and industrial interest. Recently, it has been found that the heptazine molecule, as well its substituted derivatives, exhibits a negative singlet-triplet gap, i.e. the first excited singlet state has lower energy than the first excited triplet state. This unique property, contradicing the near-universal Hund's rule, enables to harvest the energy from the triplet excitation channel and increase the quantum efficiency of the OLED devices. Our work focues on development of cost-effective quantum chemistry methods for excited state which are able to describe the singlet-triplet inversion with quantitative accuracy. For this purpose we combine the rank-reduced coupled-cluster methods, embedding techniques and density functional theory. |
|---|
Theoretical modelling of proton-matter interactions
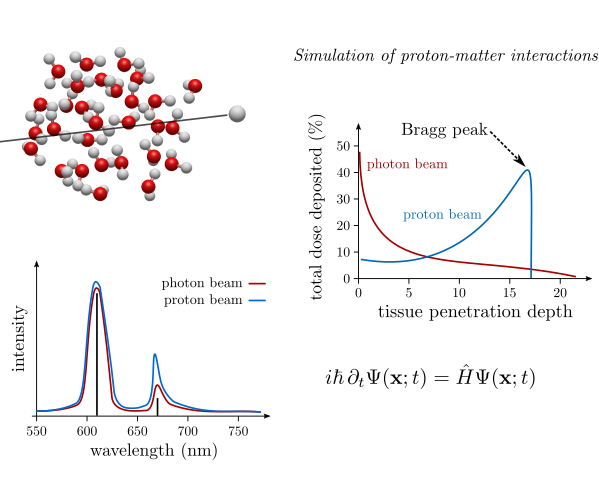 |
Accurate description of interactions between high-energy protons (as well as heavier atomic nuclei) and matter is important in many applications, in particular in radiation therapy used for treatment of various types of cancer. We are developing new electronic structure methods which are able to model such interactions, both in the perturbative regime and within explicitly time-dependent framework. These methods are not limited to description of small systems and are aimed at calculations for basic molecular structures building human tissues. We are interested, in particular, in theoretical description of several unique phenomena observed in the electronic excitation spectra, such as proton-induced fluorescence in the UV-VIS energy range or proton-induced spin-forbidden transitions. Comparison with analogous light-induced spectra enables to elucidate important differences between the two types of fluorescence and leads to better understanding of the proton-matter interactions, and discovery of novel physical phenomena. |
|---|